
L’AMILOIDOSI EREDITARIA DA TRANSTIRETINA: BASI GENETICHE E PRESENTAZIONE CLINICA
Introduzione
Le amiloidosi sistemiche sono un gruppo eterogeneo di patologie caratterizzate dal deposito extracellulare di materiale proteico costituito da fibrille insolubili, che hanno una struttura estremamente ordinata. La conformazione specifica delle fibre di amiloide è responsabile della caratteristica birifrangenza color verde mela che i tessuti infiltrati assumono dopo colorazione con rosso Congo, quando sono osservati al microscopio a luce polarizzata (Figura 1) [1].
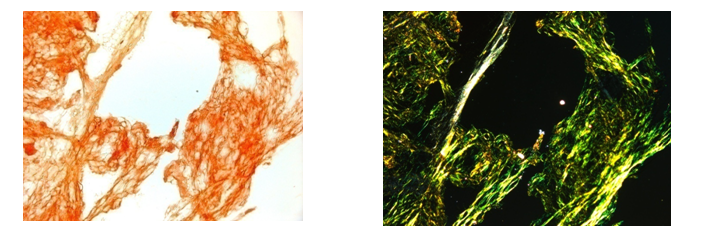 Figura 1. Aspirato di grasso periombelicale colorato con rosso Congo (a sinistra) e osservato in luce polarizzata (a destra). La caratteristica birifrangenza verde indica la presenza di amiloide nel tessuto.
Figura 1. Aspirato di grasso periombelicale colorato con rosso Congo (a sinistra) e osservato in luce polarizzata (a destra). La caratteristica birifrangenza verde indica la presenza di amiloide nel tessuto.
Sono note più di 35 proteine in grado di formare depositi di amiloide. Tuttavia, dal punto di vista clinico, le entità di maggiore interesse sono: l’amiloidosi da catene leggere (AL), l’amiloidosi associata a stati infiammatori cronici (AA) e l’amiloidosi da transtiretina (ATTR). Di questa distinguiamo la forma dovuta a mutazione del gene della transtiretina (hATTR) e la forma wild-type (ATTRwt), in passato chiamata anche amiloidosi cardiaca senile. Vi sono anche altri tipi di amiloidosi geneticamente trasmessa, molto rari e associati a mutazioni di altri geni, come lisozima, apolipoproteine A-I, A-II e C-II, e fibrinogeno.
Amiloidosi ereditaria da transtiretina (hATTR)
L’amiloidosi ereditaria da transtiretina (hATTR) è una malattia genetica rara a trasmissione autosomica dominante e a esordio in età adulta, causata da mutazioni del gene TTR. Sono note oltre 130 mutazioni di significato patologico del gene transtiretina.
La transtiretina (o prealbumina) è una proteina di trasporto sintetizzata dal fegato, dai plessi corioidei e dall’epitelio pigmentato della retina. Le sue funzioni biologiche principali sono il trasporto dell’ormone tiroideo tiroxina e il trasporto della vitamina A nel plasma attraverso il legame con un’altra proteina, chiamata retinol binding protein (RBP).
Le mutazioni di transtiretina che causano amiloidosi non compromettono le funzioni biologiche di questa proteina. Tuttavia, queste varianti promuovono importanti modificazioni strutturali e innescano una serie di eventi che determinano la formazione di aggregati amiloidi a partire da frammenti della proteina stessa. In generale, possiamo dire che quanto più una mutazione rende instabile la proteina, tanto maggiore è la rapidità con cui si generano i depositi di amiloide. Esistono anche mutazioni che aumentano la stabilità della transtiretina e queste mutazioni non causano la formazione di amiloide ma hanno al contrario un effetto di protezione.
La formazione dei primi aggregati amiloidi nei tessuti è un processo molto lento. Tuttavia, quando si generano i primi nuclei, la crescita delle fibre e dei depositi avviene con più rapidità e porta alla comparsa di iniziali segni di danno d’organo. Pertanto, la diagnosi precoce è fondamentale per intraprendere un trattamento che ostacoli l’ulteriore deposizione della proteina.
Dati epidemiologici della hATTR
La hATTR e’ una malattia presente in varie parti del mondo con incidenza variabile ed è considerata “endemica” nel nord del Portogallo e della Svezia. Foci endemici sono presenti anche in Giappone, in Brasile, nell’isola di Maiorca e a Cipro. Negli ultimi anni un numero sempre crescente di casi è stato riportato in altri paesi, quali la Germania, la Bulgaria e la Turchia.
Come detto sono state documentate oltre 130 diverse mutazioni. Soprattutto nelle zone endemiche, almeno il 50 per cento dei soggetti è portatore della mutazione Val30Met. Nei pazienti con questa mutazione la malattia si manifesta in genere più precocemente (< 50 anni) nei paesi endemici come il Portogallo e piu’ tardivamente (> 50 anni) nei paesi non endemici.
La hATTR e’ stata descritta in 36 paesi diversi con una prevalenza globale stimata di 5,000–10,000 individui nel mondo e ancora maggiore secondo uno studio recente (38,000) [2].
Una rete internazionale multicentrica, composta da esperti provenienti da 10 differenti paesi (Bulgaria, Cipro, Francia, Germania, Italia, Olanda, Portogallo, Spagna, Svezia e Turchia), ha rilevato che, anche in Europa, ad eccezione che in Bulgaria ed in Turchia, la mutazione più frequente è la Val30Met. La mutazione seconda per frequenza varia nelle diverse aree geografiche. L’Italia presenta una peculiare distribuzione geografica della malattia, con meno di un quarto dei pazienti portatori della mutazione Val30Met e con diversi focolai di altre mutazioni soprattutto nel Sud Italia, dove la malattia assume localmente caratteristiche endemiche (Figura 2) [3].
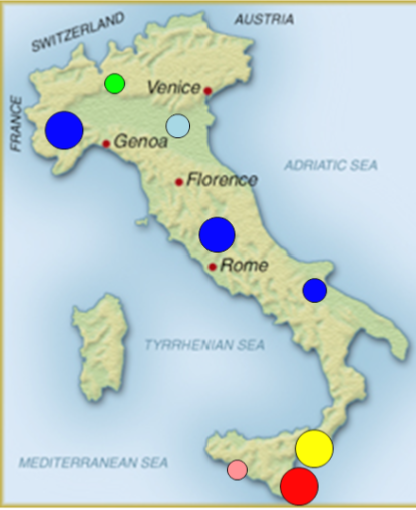
Figura 2. Distribuzione geografica delle principali mutazioni di TTR in Italia: Il colore indica la specifica mutazione responsabile della malattia in ciascun territorio. Blu: Val30Met; Azzurro: Ile68Leu; Giallo: Phe64Leu; Rosso: Glu89Gln; Verde: Tyr78Phe; Rosa: Thr49Ala.
Uno studio epidemiologico italiano è stato condotto in Sicilia, dove vi sono alcuni focolai endemici. In esso è riportata l’analisi di un gruppo di pazienti con hATTR afferiti, in un periodo di 20 anni, presso un centro di riferimento per le malattie neuromuscolari [4]. Sono stati identificati 3 fenotipi differenti, in 3 diverse aree della Sicilia, ciascuno associato a una differente mutazione. La variante Val30Met è risultata assente, mentre sono stati individuati tre foci endemici per le mutazioni Glu89Gln, Phe64Leu e Thr49Ala, con una prevalenza totale di 8,8 casi per milione di abitanti.
Registro Nazionale per l’amiloidosi ereditaria da transtiretina
Recentemente in Italia la Fondazione Telethon ha finanziato il progetto ” Registro nazionale per l’amiloidosi familiare da transtiretina: Rete collaborativa multicentrica per la definizione della storia naturale, degli standard di cura, del carico e dei bisogni dei pazienti (http://www.registronmd.it)”.
Lo studio si è svolto attraverso la creazione di una Registro Nazionale on-line per la hATTR, dove sono raccolte in forma anonima le informazioni cliniche e genetiche di tutti i pazienti italiani che hanno aderito (http://www.registronmd.it).
I risultati dello studio stanno fornendo importanti dati epidemiologici sulla prevalenza italiana della malattia, sulla storia naturale, sugli standard di cura, sul carico e sui bisogni dei pazienti e faciliteranno la fattibilità e la progettazione di futuri studi terapeutici. I primi dati sono stati recentemente pubblicati [5]. Lo studio, coordinato dal prof. Giuseppe Vita dell’Università di Messina, include numerosi centri italiani di riferimento per le neuropatie periferiche e per l’amiloidosi. Il Registro Italiano ha l’obiettivo ambizioso di includere tutti i pazienti e i portatori presintomatici presenti sul territorio nazionale.
Clinica della hATTR
La hATTR è una malattia sistemica e può pertanto coinvolgere diversi organi, con decorso progressivo e potenzialmente fatale se non diagnosticata precocemente. Le caratteristiche cliniche possono variare in relazione al tipo di mutazione e agli organi coinvolti. L’amiloidosi ereditaria da transtiretina si manifesta prevalentemente con sintomi e segni clinici di coinvolgimento del sistema nervoso periferico (SNP), del sistema nervoso autonomo (SNA) e del cuore. Il coinvolgimento renale (proteinuria, insufficienza renale) e i sintomi oculari (opacità del vitreo, glaucoma, anomalie pupillari) sono anch’essi frequenti nei pazienti con amiloidosi da mutazioni TTR.
La presentazione tipica della hATTR è una neuropatia sensitivo-motoria di tipo assonale. I sintomi classici includono alterazioni della sensibilità al dolore e alla temperatura, “bruciore”, “formicolio e intorpidimento” agli arti, dolore neuropatico, riduzione della forza, debolezza muscolare, difficoltà nella deambulazione e disturbi dell’equilibrio. L’interessamento del sistema nervoso autonomo comporta alterazioni della pressione arteriosa (talora con sincopi), tachicardia, ipotensione ortostatica, impotenza, alvo alterno (stipsi/diarrea) e alterazioni della sudorazione.
Molti pazienti lamentano precocemente perdita dell’appetito e calo del peso corporeo. Altre manifestazioni a carico dell’apparato gastrointestinale includono nausea, vomito e senso di sazietà precoce.
La sindrome del tunnel carpale, sebbene non sia specifica della hATTR, è una manifestazione precoce e può esordire molto prima degli altri sintomi neurologici, anticipando la diagnosi di hATTR anche di dieci anni. Analogamente, una stenosi spinale lombare può essere un segno precoce. La cardiomiopatia amiloidosica è caratterizzata da ispessimento delle pareti del cuore, scompenso cardiaco, disturbi del ritmo e blocchi di conduzione. Le manifestazioni più comuni legate al coinvolgimento del cuore comprendono comparsa di gonfiore alle gambe (edemi), mancanza di fiato, facile faticabilità [6].
Molto raramente, alcune mutazioni possono associarsi a coinvolgimento del sistema nervoso centrale (crisi epilettiche, attacchi ischemici transitori, ictus). Alla luce di quanto detto, la gestione clinica e terapeutica deve avvalersi di diversi specialisti ed è pertanto importante che ogni paziente sia seguito da un team multidisciplinare.
Diagnosi
Per formulare la diagnosi di hATTR è necessario eseguire il test genetico e dimostrare la presenza di una mutazione amiloidogenica del gene della transtiretina. Parallelamente, si deve ricercare la presenza di depositi di amiloide mediante una biopsia non invasiva, come l’aspirato di grasso periombelicale o la biopsia delle ghiandole salivari minori labiali. L’identificazione di un tessuto positivo consente infatti la caratterizzazione delle fibre di amiloide, confermando la presenza di transtiretina nei depositi ed escludendo altre forme clinicamente simili di amiloidosi.
E’ quindi importante determinare il coinvolgimento d’organo mediante un’accurata valutazione clinica e alcuni esami diagnostici specifici. Per l’interessamento cardiaco, oltre all’ecocardiografia, alla risonanza magnetica e ai biomarcatori, è particolarmente utile l’esecuzione di una scintigrafia con tracciante osseo, che permette di distinguere l’amiloidosi da transtiretina da altre forme di cardiopatia ipertrofica.
Vi è oggi unanime consenso sul fatto che la presenza di captazione cardiaca del tracciante alla scintigrafia ossea consenta di formulare la diagnosi di amiloidosi da transtiretina anche senza conferma istologica, purché sia esclusa una concomitante amiloidosi AL sulla base di una immunofissazione del siero e delle urine negativa e di un rapporto k/λ delle catene leggere libere sieriche nella norma. In questi casi il test genetico deve completare la diagnosi per distinguere una forma geneticamente trasmessa dalla più frequente forma acquisita causata dalla transtiretina non mutata o wild-type. Questa forma è più frequentemente osservata nella popolazione maschile a partire dall’età di 60 anni.
Bibliografia:
1. Merlini G and Bellotti V. Molecular Mechanisms of Amyloidosis. New Engl J Med, 2003.
2. Schmidt HH et al. Estimating the Global Prevalence of Transthyretin Familial Amyloid Polyneuropathy. Muscle Nerve, 2018.
3. Parman Y et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol, 2016.
4. Mazzeo A et al. Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP): A Single-Center Experience in Sicily, an Italian Endemic Area. J Neuromuscul Dis, 2015.
5. Russo M et al. ATTRv amyloidosis Italian Registry: clinical and epidemiological data. Amyloid 2020.
6. Rapezzi C et al. Disease Profile and Differential Diagnosis of Hereditary Transthyretin-Related Amyloidosis With Exclusively Cardiac Phenotype: An Italian Perspective. Eur Heart J, 2013