
L’amiloidosi era storicamente considerata una patologia rara e poco conosciuta. Grazie però alla florida letteratura scientifica e alla notevole semplificazione dell’approccio diagnostico la prevalenza della patologia sta rapidamente cambiando. Numerosi aspetti della malattia sono stati esplorati in termini di manifestazioni cliniche, caratteristiche strumentali e di stratificazione prognostica.
Le amiloidosi sono un gruppo eterogeneo di patologie acquisite o ereditarie, localizzate o sistemiche, che condividono una caratteristica: la deposizione extracellulare di proteine fibrillari insolubili che determina una disorganizzazione della struttura dei tessuti coinvolti con conseguente disfunzione d’organo [1,2].
Nonostante le proteine implicate nel processo amiloidogenico siano diverse a seconda del tipo di amiloidosi, esse condividono proprietà tintoriali e strutturali comuni [1-4]:
• un aspetto di sostanza amorfa al microscopio a luce normale (Figura 1, sinistra);
• la birifrangenza “apple green” con la colorazione Rosso Congo al microscopio a luce polarizzata (Figura 1, destra);
• un’ultrastruttura composta da fibrille di diametro variabile tra 7 e 10 nm, costituite da amiloide e da altri fattori additivi, quali i proteoglicani;
• una conformazione “β pleated-sheet” alla spettroscopia a raggi infrarossi, determinante principale del potenziale amiloidogenico della proteina.
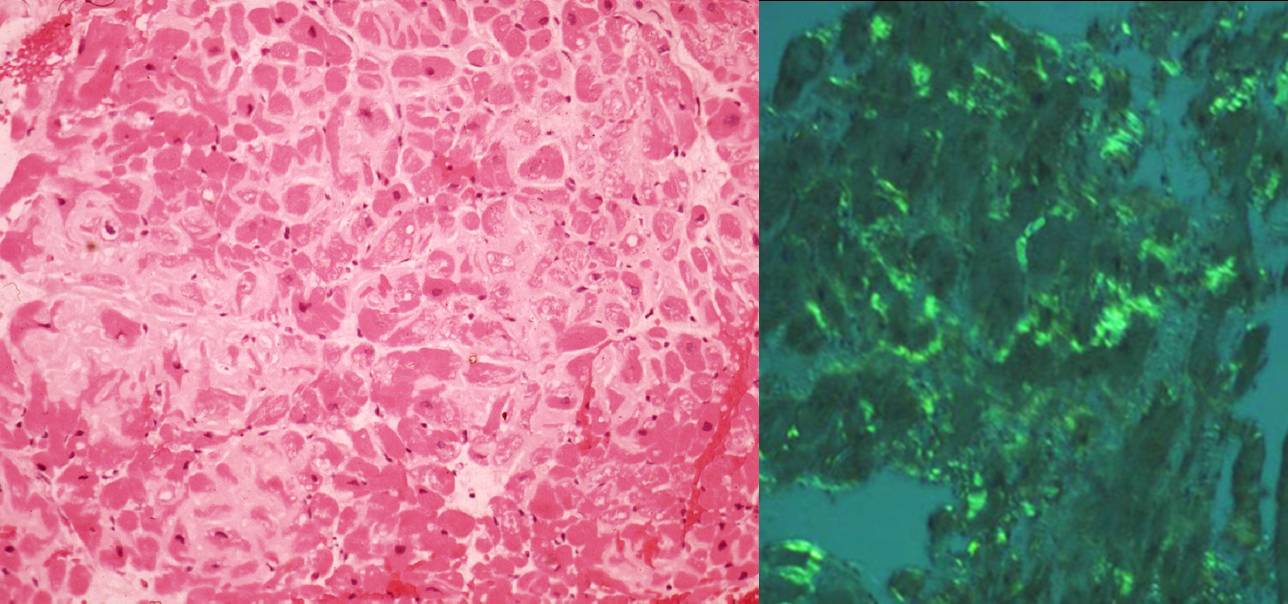
Figura 1. Rilievi istologici tipici di miocardio infiltrato da sostanza amiloide: nel riquadro a sinistra i depositi di amiloide (in rosa chiaro dopo colorazione con Ematossilina-Eosina) hanno la caratteristica di una sostanza omogenea ed eosinofila che infiltra diffusamente il tessuto miocardico, isolando anatomicamente e funzionalmente singole cellule o gruppi di cellule. La tipica birifrangenza verde mela (nel riquadro a destra) si osserva al microscopio a luce polarizzata dopo colorazione con rosso Congo.
In generale il cuore è uno degli organi “bersaglio” in cui più frequentemente si deposita l’amiloide: in alcune forme è la principale causa di morbilità e mortalità dei pazienti, in altre può invece rappresentare un reperto accidentale e privo di significato funzionale [1,2].
L’amiloidosi cardiaca non rappresenta una singola entità, ma è caratterizzata da un background patogenetico eterogeneo riconducibile, nel mondo occidentale, principalmente a tre forme eziologiche distinte [5,6,7]:
1) l’amiloidosi AL (un tempo nota come amiloidosi primaria) secondaria alla presenza di cloni plasmacellulari nel midollo osseo che producono catene leggere libere circolanti delle immunoglobuline responsabili dei depositi fibrillari;
2) l’amiloidosi ereditaria correlata alla transtiretina (ATTR), malattia autosomica dominante con espressività variabile e penetranza incompleta, che può essere causata da oltre 100 mutazioni del gene della transtiretina;
3) l’amiloidosi sistemica senile (Senile Systemic Amyloidosis, SSA) da depositi di transtiretina non mutata (wild-type).
Inoltre, come conseguenza di infezioni croniche, nei Paesi in via di sviluppo non è infrequente l’amiloidosi secondaria (AA) [5].
Infine, ci sono rare forme ereditarie non transtiretino-correlate quali quelle secondarie a mutazioni per il fibrinogeno, l’apolipoproteina (Apo-AI ed Apo-AII) e la gelsolina [5]. Le mutazioni per il fibrinogeno e l’apolipoproteina determinano principalmente nefropatie progressive e solo raramente causano cardiomiopatie. La mutazione della gelsolina invece, endemica in Finlandia ma estremamente rara nel resto del mondo, provoca principalmente neuropatie, nefropatie e a livello cardiaco si manifesta pressoché esclusivamente con disturbi della conduzione [5].
La cardiomiopatia amiloidotica è considerata lo stereotipo delle cardiomiopatie infiltrative ed è caratterizzata da un incremento degli spessori parietali con fisiologia restrittiva ed aumentata rigidità strutturale che causa un rapido incremento delle pressioni intraventricolari in associazione ad un solo lieve incremento dei volumi di riempimento [8,9].
Oltre al coinvolgimento parietale, l’amiloide infiltra gli atri (favorendo la comparsa di aritmie sopraventricolari e la formazione di trombi), i vasi intramiocardici (determinando ischemia miocardica), ed il sistema di conduzione (determinando blocchi atrioventricolari ed intraventricolari, talora con necessità di impianto di pace-maker) [1].
A causa della natura proteiforme della malattia, l’elemento centrale per la diagnosi di cardiomiopatia amiloidotica è rappresentato dal sospetto. Gli strumenti principali per un sospetto di amiloidosi cardiaca sono l’elettrocardiogramma (ECG) e l’ecocardiogramma e in particolare la lettura integrata delle due metodiche.
Un ulteriore esame diagnostico a disposizione è rappresentato dalla scintigrafia total body con tracciante osseo. Nel corso degli ultimi decenni sono stati studiati diversi traccianti scintigrafici (principalmente fosfonati) per la valutazione dei depositi di amiloide a livello cardiaco, con risultati molto eterogenei e talora contraddittori. Nei pazienti affetti da cardiomiopatia amiloidotica transtiretino-correlata (sia mutata sia wild-type) la scintigrafia total body mostra una captazione del tracciante osseo a livello cardiaco secondo un grado (“Perugini score” o “Bologna score”) da 2 a 3. Tale indagine permette di porre diagnosi differenziale (con alcune accezioni) con l’amiloidosi AL in cui non si osserva captazione del medesimo tracciante. [16].
Sebbene le metodiche diagnostiche a disposizione abbiano condotto alla possibilità di giungere alla diagnosi non invasiva della malattia, l’analisi istologica resta comunque il gold standard diagnostico, con la dimostrazione in almeno un organo coinvolto della caratteristica birifrangenza “apple green” al microscopio a luce polarizzata, dopo fissazione del prelievo con il colorante rosso Congo [17].
L’eziologia finale di amiloidosi cardiaca è determinata dall’indagine immunoistochimica (con anticorpi specifici) sul reperto bioptico [15] sebbene moderne tecniche di proteomica siano usate sempre più di frequente poiché più specifiche e sensibili [18].
L’analisi genetica permetterà di identificare mutazioni a carico del gene della transtiretina nelle forme familiari.
Amiloidosi correlata alla transtiretina
La transtiretina è una proteina in grado di formare le fibrille di amiloide in vivo ed è associata a due forme distinte di cardiomiopatia amiloidotica: 1) la forma transtiretino-relata ereditaria (ATTR) causata da mutazioni puntiformi del gene per la transtiretina; 2) l’amiloidosi sistemica senile (SSA) forma non ereditaria associata a depositi di transtiretina non mutata (wild-type) [19].
L’amiloidosi transtiretino-relata ereditaria (ATTR) è la forma più frequente di amiloidosi sistemica familiare.
La transtiretina umana (TTR) è una proteina sierica deputata al trasporto del RBP (proteina che lega il retinolo o vitamina A); viene prodotta principalmente dal fegato ed in piccola parte dal plesso coroideo e dalla retina. La TTR (Figura 4) circola come omotetramero composto da 4 subunità identiche legate in modo non-covalente. Ogni monomero è costituito da 127 aminoacidi ordinati in 8 domini antiparalleli a beta-foglietto [20].
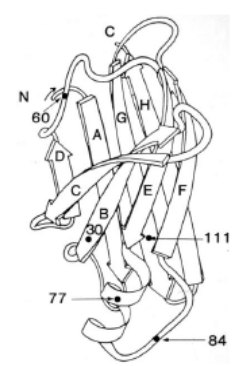
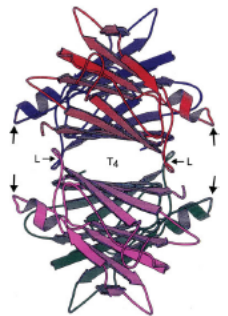
Figura 4. A sinistra il diagramma del monomero di TTR che mostra alcuni siti di mutazioni patologiche. N: sito amino terminale dei 127 aa; C: sito carbossiterminale. Le punte di freccia indicano la direzione dall’N al C degli 8 beta-foglietti antiparalleli su due piani DAGH e CBEF. A destra il diagramma del tetramero in cui ogni monomero è rappresentato da un colore diverso. Le frecce più grandi indicano il sito di legame per il retinolo/vitamina A.
Il gene che codifica per la TTR è collocato sul cromosoma 18 ed è costituito da 4 esoni e 5 introni. Sono state identificate circa 100 mutazioni geniche da cui derivano altrettante varianti proteiche. La modalità di trasmissione è autosomica dominante e la maggior parte degli individui affetti è eterozigote per la mutazione patogena [20]. La penetranza di questa patologia è estremamente variabile e la stessa mutazione può fenotipicamente presentarsi in maniera differente tra i vari componenti della stessa famiglia o addirittura non manifestarsi (portatori sani) [1].
Per molto tempo si è ritenuto che la malattia fosse conseguenza della sola mutazione Val30Met (sostituzione di una Valina con una Metionina in posizione 30 della catena polipeptidica), in quanto le prime segnalazioni ed i casi studiati derivavano da focolai giapponesi [21], svedesi [22] e portoghesi [23], aree in cui tale mutazione risulta endemica.
La prevalente estrinsecazione neurologica della malattia nelle forme endemiche correlate alla mutazione Val30Met ha fatto si che l’ATTR fosse per lungo tempo considerata una malattia di pertinenza neurologica, tanto da essere comunemente chiamata Polineuropatia Amiloidotica Familiare (FAP). La successiva segnalazione di nuove mutazioni della TTR [24-28] (spesso confinate a piccole e limitate aree geografiche) ha rilevato l’esistenza di una eterogeneità genotipica a cui corrisponde un ampio spettro di manifestazioni cliniche, nell’ambito della quale si inserisce anche il coinvolgimento cardiaco. Questa eterogeneità è correlata a diversi fattori quali le specifiche mutazioni, l’età di esordio della malattia [29,30], il sesso dei pazienti (parziale protezione del sesso femminile, almeno fino alla menopausa) [31,32], del genitore responsabile della trasmissione del gene mutato (malattia più precoce e grave se trasmessa dalla madre), la composizione delle fibrille (transtiretina intera o frammentata [33]), la distribuzione geografica e la tipologia di aggregazione: endemica o non endemica [19,21,22,29,32,34].
Nella forma endemica “classica” Portoghese e Giapponese la patologia è caratterizzata da una polineuropatia sensitivo-motoria ad alta penetranza (almeno 80%), che colpisce egualmente maschi e femmine, che si manifesta intorno ai 30-35 anni (mostrando il fenomeno dell’anticipazione generazionale) con una disfunzione delle fibre periferiche degli arti inferiori che poi si estende alle estremità prossimali e che raggiunge lo stadio terminale in 10-15 anni [27,35].
La forma endemica Svedese invece è più lenta, tardiva e con bassa penetranza (circa il 5%) [35] e nonostante preveda analoga mutazione non sembra avere un fondatore comune alle altre due [34,36]. Inoltre, è emersa l’esistenza di due distinte forme svedesi Val30Met correlate (Tipo A e tipo B), a seconda della composizione fibrillare di TTR, troncata o intera [37].
Le manifestazioni cliniche della forma endemica classica sono tipicamente disturbi della sensibilità termica e dolorifica, parestesie e disestesie agli arti inferiori. La funzione motoria viene interessata nelle fasi avanzate e si estrinseca con importante disabilità [19,38]. Precocemente si manifesta anche una disautonomia progressiva dovuta sia a denervazione simpatica che parasimpatica, caratterizzata da turbe della motilità gastrointestinale (costipazione alternata a diarrea che determina perdita di peso e malnutrizione), ritenzione o incontinenza urinaria, disfunzione erettile ed ipotensione ortostatica [20,21]. L’interessamento cardiaco prevede quasi esclusivamente disturbi della conduzione talora tali da richiedere l’impianto di pace-maker. Le cardiomiopatie sono invece rarissime e, nel caso, tardive [21]. Presenti infiltrati renali, anche consistenti (identificazione post-mortem) ma con funzione d’organo del tutto conservata fino alla fine [21].
Il coinvolgimento miocardico nella ATTR può risultare solo un componente di un complesso quadro principalmente neurologico o rappresentare l’unica espressione di malattia. E’ questo il caso della mutazione Val122Ile, probabilmente la mutazione più frequente dal momento che ha una prevalenza di circa il 4% nella popolazione Afro-Americana, caratterizzata da severa cardiomiopatia restrittiva ad insorgenza tardiva (VI-VII decade) senza o con minimo interessamento neurologico [39-42]; altre mutazioni presentano un importante, talora dominante o esclusivo, interessamento cardiaco: la mutazione Thr60Ala Irlandese (contea di Donegal) e degli Appalachi con cardiomiopatia restrittiva ad insorgenza tardiva ed interessamento neurologico [43,44]; la mutazione Leu111Met Danese [22,45] caratterizzata da cardiomiopatia ad insorgenza precoce (30-40 anni) e neuropatia non sempre manifesta.
Proprio in relazione alla vasta eterogeneità, anche fenotipica, il clinico può quindi trovarsi di fronte a casi con un esclusivo interessamento neurologico nell’ambito di una chiara familiarità o a casi in cui è presente un quadro clinico dominato da un importante coinvolgimento miocardico ed interessamento neurologico assente o molto sfumato. Questo ampio spettro di presentazioni cliniche rende il riconoscimento di ATTR particolarmente difficile, soprattutto in ambito cardiologico.
La sindrome del tunnel carpale si associa frequentemente all’amiloidosi TTR-relata e, per motivo non ancora ben noto, precedendo la clinica cardiologica di circa 10 anni. [22]. Si tratta di una patologia comune nella popolazione generale [46], caratterizzata dall’intrappolamento dei tendini del polso con conseguente disfunzione del nervo mediano.
La transtiretina non mutata (wild type) è la proteina precursore dell’amiloidosi sistemica senile (SSA). La SSA è una patologia che interessa molto spesso uomini di età superiore a 65 anni e determina un coinvolgimento sistemico (polmoni, tratto gastroenterico, fegato, milza, ghiandole endocrine) anche se, dal punto di vista clinico, le uniche due manifestazioni sono la cardiomiopatia ed il tunnel carpale che in genere si presenta 3-5 anni prima dei sintomi cardiologici [1,5]. Nei pazienti anziani la transtiretina pur strutturalmente normale può diventare instabile e costituire degli aggregati mal ripiegati che si aggregano e precipitano sottoforma di amiloide [47,48].
BIBLIOGRAFIA
1. Falk RH, Dubrey S.W. Amyloid Heart disease. Prog Cardiovasc Dis 2010;52:347–361.
2. Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med 2006;166:1805–1813.
3. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid 2010;17:101–104.
4. Merlini G, Bellotti V. Molecular Mechanisms of Amyloidosis. NEJM 2003; 349:583-96.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis and referral. Heart 2011; 97: 75-84.
6. Rapezzi C, Merlini G, Quarta CC, et al. Systemic Cardiac Amyloidoses. Disease Profiles and Clinical Courses of the 3 Main Types. Circulation 2009;120:1203–1212.
7. Merlini G, Seldin Davide C., Gertz Morie A. Amyloidosis: Pathogenesis and New Therapeutic Options. J Clin Oncol 2011; 29: 1924-1933.
8. Maron BJ, Towbin JA, Thiene G, et al. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement From the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006;113:1807–1816.
9. Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2008;29:270–276.
10. Murtagh B, Hammil SC, Gertz MA, et al. Electrocardiographic findings in primary sistemic amyloidosis and biopsy –proven cardiac involvement. Am J Cardiol 2005; 95: 535-7.
11. Dubrey SW., Cha K, Anderson J et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. Q J Med 1998;91:141-57.
12. Rapezzi C, Riva L, Quarta CC, et al. I trabocchetti e gli inganni dell’amiloidosi cardiaca. G Ital Cardiol 2007; 8 (6): 377-38.
13. Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol 1982;49:9–13.
14. Rahman JE, Helou EF, Gelzer-Bell R et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol 2004;43:410-415
15. Quarta CC, Borghi C, Perlini S et al. A simple voltage/mass index improves diagnosis of cardiac amyloidosis in patients with unexplained left ventricular “hypertrophy”: an electrocardiographic and echocardiographic study of more than 500 patients. Circulation 2010;122(21):Abstact 16852.
16. Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005;46:1076–1084.
17. Arbustini E, Verga L, Concardi M et al. Amyloid: J Protein Folding Disord 2002; 9:108-114.
18. Stoppini M, Obici L, Lavatelli F, et al. Proteomics in protein misfolding disease. Clin Chem Lab Med 2009; 47: 627-635.
19. Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol 2010;7:398–408.
20. Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle & Nerve 2007; 36: 411-423.
21. Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology. 2002; 58:1001-1007.
22. Suhr OB, Svendsen IH, Andersson R et al. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med 2003; 254:225-235.
23. Coelho T, Sousa A, Lourenco E et al. A study of 159 Portoguese patients with familial amyloidotic polyneuropathy (FAP) whose parents were both unaffected. J Med Genet 1994;31:293-299.
24. Plante-Bordeneuve V, Carayol J, Ferreira A et al. Genetic study of transthyretin amyloid neuropathies : carrier risk among French and Portuguese families. J Med Genet 2003;40:120.
25. Plante-Bordeneuve V, Ferreira A, Lalu T et al. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology 2007; 693-698.
26. Adams D, Reilly M, Harding AE et al. Demonstration of genetic mutation in most of the amyloid neuropathies with sporadic occurrence. Rev Neurol 1992; 148:736-741.
27. Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol 2005;62:1057-1062.
28. Rapezzi C, Perugini E, Salvi F et al. Phenotypic and genotipic heterogeneity in transthyretin-related cardiac amyloidosis: towards tailoring of therapeutic strategies? Amyloid 2006; 13 (3): 143-153.
29. Conceição I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late- and early-onset cases in Portugal. Muscle Nerve 2007; 35:116-118.
30. Koike H, Misu K, Ikeda S et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early vs late-onset form. Arch Neurol 2002; 59: 1771-6.
31. Rapezzi C, Riva L, Quarta CC, et al. Gender related risk of myocardial involvement in sistemi amyloidosis. Amyloid 2008; 15 (1):
40-48.
32. Bittencourt PL, Couto CA, Clemente C et al. Phenotypic expression of familial amyloid polyneuropathy in Brazil. Eur J Neurol 2005; 12: 289-293.
33. Bergstrom J, Gustavsson A, Hellman U et al. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. JPathol 2005; 206: 224-232.
34. Ohmori H, Ando Y, Makita Y et al. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J Med Genet 2004; 41:51-55.
35. Soares ML, Coelho T, Sousa A et al. Haplotypes and DNA sequence variation within and surrounding the transthyrettin gene: genotype-phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur J of Human Genetics 2004; 12:225-237.
36. Drugges U, Andersson R, Chizari F et al. Familial amyloidotic polineuropathy in Sweden: a pedigree analysis. J Med Genet 1993; 30: 388-392.
37. Okamoto S, Zhao Y, Lindqvist P et al. Development of cardiomyopathy after liver transplantation in Swedish hereditary tranthyretin amyloidosis (ATTR) patients. Amyloid 2011; 18 (4): 200-205.
38. Gasperini RJ and Small DH. Neurodegeneration in Familial Amyloidotic Polyneuropathy approved for publication in the Clinical and Experimental Pharmacology and Physiology. Clin Exp Pharmacol Physiol. 2011 [Epub ahead of print].
39. Jacobson DR, Pastore R, Malendowics SPS et. al. Revised transthyretin Ile 122 allele frequency in African Americans. Hum Genet 1996; 98: 236-238.
40. Jacobson DR, Pastore RD, Yaghoubian R et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. NEJM 1997;336:466-473.
41. Jacobson D, Tagoe C, Schwartzbard A et al. Relation of clinical, echocardiographic and electrocardiographic features of cardiac amyloidosis to the presence of the transthyretin V122I allele in older African-American men. Am J Cardiol 2011; 108:440-444.
42. Dharmarajan K, Maurer MS. Transthyretin Cardiac Amyloidoses in Older North Americans. J Am Geriatr Soc 2012.
43. Benson MD, Wallace MR, Tejada E et al. Hereditary Amyloidosis: Description of a new American kindred with late onset cardiomiopathy. Appalachian Amyloid. Arthritis and Rheumatism 1987; (30) 2: 195-200.
44. Sattianayagam PT, Hahn AF, Whelan CJ et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J 2011 [Epub ahead of print, doi:10.1093/eurheartj/ehr383].
45. Ranløv I, Alves IL, Ranløv PJ et al. A Danish kindred with familial amyloid cardiomyopathy revisited: identification of a mutant transthyretin-methionine111 variant in serum from patients and carriers. Am J Med 1992;93:3-8.
46. Atroshi I, Gummesson C, Johnsson R et al. Prevalence of carpal tunnel syndrome in a general population. JAMA 1999; 282: 153-158.
47. Cornwell GG III, Murdock WL, Kyle RA et al. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med 1983; 75: 618-23.
48. Belinda Ng, Connors LH, Davidoff R et al. Senile systemic amyloidosis presenting with heart failure. A comparison with light chain-associated amyloidosis. Arch Intern Med 2005; 165: 1425-1429.